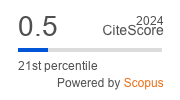
INTERNAL MEDICINE
Alport syndrome (AS) is a hereditary nephropathy caused by mutations in the COL4A3, COL4A4, and COL4A5 genes. Rare contiguous COL4A5–COL4A6 alterations cause AS with diffuse leiomyomatosis (AS-DL).
Case report. A 16-year-old male had mild proteinuria, hematuria, hearing loss and myopia since childhood. Estimated glomerular filtration rate was 82.8 mL/min/1.73 m2. Kidney biopsy showed segmental mesangial sclerosis; immunofluorescence was negative. Electron microscopy demonstrated diffuse glomerular basement membrane thinning and podocyte foot-process effacement. Two deceased brothers had end stage kidney disease; the mother had hematuria, uterine myoma, and a benign bladder tumor. A diagnosis of X-linked AS was established according to the Flinter criteria and nephroprotective treatment was initiated. Since 2024, the patient had complained of epigastric symptoms. An endoscopy revealed a 2-cm gastric submucosal lesion. A whole-exome sequencing identified a hemizygous missense variant in COL4A5 and a COL4A6 exon 1–2 deletion, confirming AS-DL.
Discussion. This case demonstrates the co-occurrence of AS-DL and shows that early pedigree assessment, combined with integrated clinicopathologic-genetic evaluation in a multidisciplinary framework, enables a timely diagnosis of atypical familial AS-DL and improves clinical management.

ISSN 2658-3348 (Online)